

We are interested in the mechanisms by which genes are transcriptionally regulated and how these processes can malfunction to cause disease. All genes are packaged in the nucleus as chromosomes (or "chromatin") by associating with small basic proteins called "histones" and other factors. Many different types of chromatin structures can form on individual genes and it is the nature of these structures that determines whether a gene will be active or inactive in a particular tissue. In many diseases, there are significant changes in the expression of specific genes either by aberrant activation or repression that are often accompanied by targeted alterations in chromatin structure. Thus, the regulation of chromatin structure is critical for proper genomic programming which impacts gene expression, DNA repair, chromosomal stability, and maintenance of tissue differentiation and cellular function. Elegant mechanisms have been defined that modulate chromatin structure to allow interaction with proteins that establish proper patterns of gene expression in specific tissues. These mechanisms involve large enzymatic protein complexes that structurally disrupt or chemically modify histone-DNA contacts within nucleosomes to facilitate gene transcription or silencing. The critical issue is how such diverse chromatin "remodeling or modifying" complexes are selectively targeted to individual genes in a variety of tissues during differentiation and under conditions that lead to disease.
We have directed our efforts towards understanding how transcriptional regulation is achieved through chromatin using genes that are controlled by very distinct processes: developmental regulation and tumorigenesis. The human ß-globin gene family is an important paradigm of mammalian tissue-specific and developmental gene regulation. Individual genes within this family are sequentially expressed during embryonic, fetal, and adult stages of erythroid differentiation primarily by the recruitment of erythroid-restricted proteins to specific promoter regions. Our studies reveal that activation of the adult ß-globin gene by the zinc finger-containing transcription factor EKLF is achieved in combination with a specific mammalian chromatin remodeling complex, SWI/SNF. SWI/SNF is a large multi-subunit protein complex that has the general property of disrupting the structure of nucleosomes. This interesting complex exists in biochemically distinct forms and specific subunits are required for normal development and tumor suppression. We have demonstrated that SWI/SNF shows selectivity in the types of transcription factors that it functions with and the genes that it regulates. Using recombinant proteins and chromatin-assembled genes, we demonstrate that only two subunits of SWI/SNF are required to interact with zinc finger DNA-binding proteins to catalyze targeted interaction with chromatinized genes. Moreover, we observe functional specificity among different types of SWI/SNF complexes. We are extending this analysis to address the larger issue of how chromatin remodeling/modifying complexes are directed towards specific gene programs during the transition between tissue proliferation and differentiation.
To analyze gene regulation during tumorigenesis, we have focused on the tumor suppressor protein, p53. p53 is a DNA-binding transcription factor that controls distinct programs of gene expression in response to DNA damage and cellular stress. The importance of this protein is that it is mutated in the majority of all human cancers thus impairing the normal processes of cell cycle arrest and apoptosis. To decipher how p53 regulates these critical processes to prevent malignancy, we have conducted biochemical and cell-based analyses that reveal distinct transcriptional mechanisms utilized by this tumor suppressor to determine cell fate. Our studies reveal that, far from being a latent tumor suppressor, p53 functions in a temporal manner to regulate promoter activity both before and after cellular stress. This is achieved by the ability of p53 to establish markedly different affinities of RNA polymerase II (RNAP II) on its diverse target promoters and recruit transcriptional initiation components in a stress-specific manner. In this way, cell cycle arrest genes are rapidly activated after DNA damage by conversion of high levels of bound RNAP II to an elongating form. By contrast, apoptotic genes have low levels of bound RNAP II but undergo stress-induced activation through efficient re-initiation. The behavior of the RNAP II complex determines the kinetics of expression of individual genes within the p53-regulated program which, in turn, controls the choice between cell cycle arrest or apoptosis. We are currently examining how p53 directs the recruitment of RNAP II and specific cofactors to its diverse target promoters before and after stress to generate the appropriate transcriptional response and how this fails in human cancers.
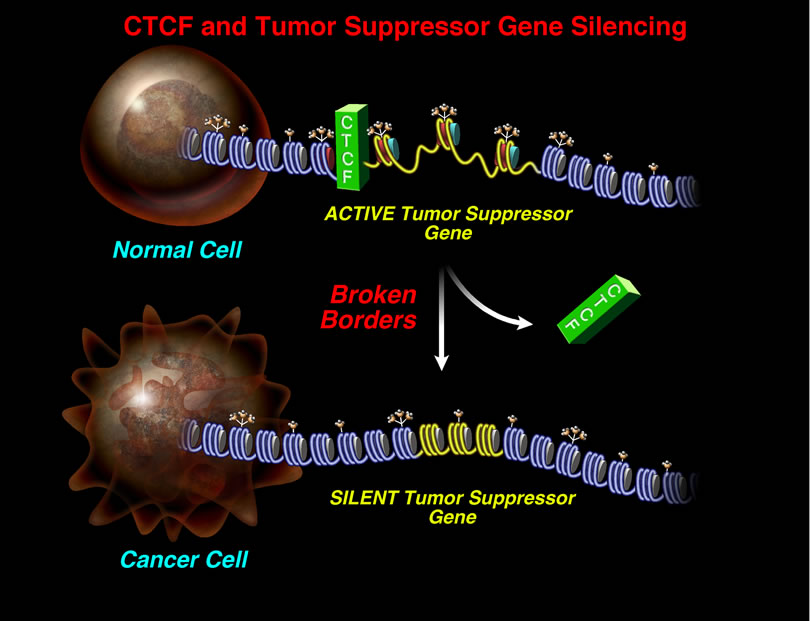
Global regulation of transcription occurs through epigenetic changes that are catalyzed by diverse chromatin enzymatic complexes. We are identifying which chromatin complexes are recruited to specific genes and determining the mechanism by which they are selectively targeted to establish programmed patterns of gene expression. These issues are being explored using embryonic stem cells, to analyze normal mechanisms of gene programming that regulate cell fate; and during early stages of tumorigenesis, to examine how these mechanisms go awry to initiate genomic instability. For example, gene silencing by DNA methylation is required for normal cellular differentiation, however, inappropriate silencing of critical genes by DNA methylation also occurs in a variety of human cancers. By understanding how chromatin structure and function is normally modulated by diverse enzymatic complexes, we hope to gain insight into how these complexes are mistargeted during disease.